A sobrevida em 5 anos da amiloidose de cadeias leves (AL), anteriormente de 15%, agora se aproxima dos 50%, em grande parte às custas de realocação do arsenal terapêutico destinado, inicialmente, ao mieloma múltiplo. A despeito da estabilidade da incidência, com média de 12 casos por milhão de pessoas-ano, a prevalência tem aumentado progressivamente, acompanhando o envelhecimento populacional, migrando de 15 para 40 casos por milhão entre 2007 e 2015, conforme dados norte-americanos.

O que é a amiloidose?
A amiloidose tem origem no desdobramento incorreto da estrutura tridimensional de um precursor proteico solúvel (tendo sido descritos mais de 40!), gerando agregação de oligômeros em forma de fibrilas amiloides beta-pregueadas e com arranjo não paralelo.
A agregação de compostos proteicos resulta em depósito amiloide no espaço extracelular de vários órgãos e tecidos, gerando efeitos compressivos locais e citotoxicidade, desencadeando disfunções orgânicas progressivas e mortalidade.
O termo amiloide foi cunhado pela sua semelhança histológica ao amido após a coloração por iodo. Entretanto, a estrutura dos precursores é proteica, e não polissacarídica.
Quais as formas mais comuns de amiloidose?
As duas principais formas de amiloidose são adquiridas: a amiloidose AL e a amiloidose transtirretina selvagem (ATTRwt), sendo a última a mais prevalente, especialmente entre homens idosos e com acometimento predominantemente cardíaco.
O que é amiloidose AL?
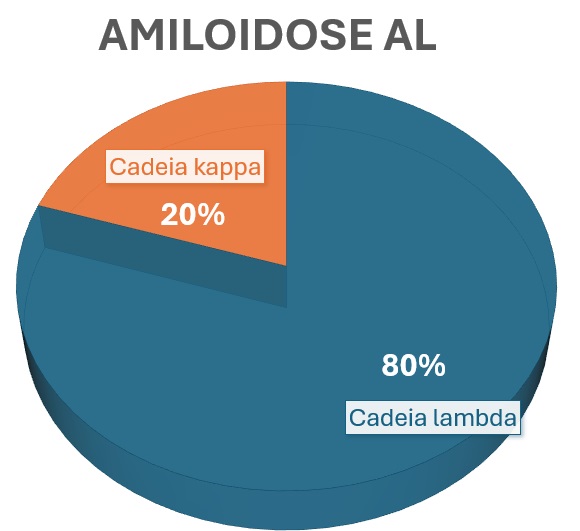
A amiloidose AL, por sua vez, é uma discrasia de plasmócitos clonais, capazes de produzir cadeias leves de imunoglobulinas de forma anormal e excessiva, prioritariamente do tipo lambda, com potencial amiloidogênico.
As cadeias leves anormais, ao interagirem com os glicosaminoglicanos e proteína P amiloide sérica, formam agregados amiloides.
Um marco característico da amiloidose AL é que, em até metade dos casos, há uma translocação cromossômica: t(11;14).
Quais são os principais fatores de risco para a amiloidose AL?
Os principais fatores de risco para a amiloidose AL são as gamopatias monoclonais pré-existentes e o mieloma múltiplo. O risco relativo de amiloidose AL na presença de gamopatia monoclonal de significado indeterminado (MGUS) é de 8,8% e de até 15% entre os pacientes com mieloma múltiplo. O aumento das cadeias livres séricas monoclonais geralmente precede o desenvolvimento da amiloidose AL em mais de 4 anos.
Quais são as suas principais apresentações clínicas?
A sintomatologia inicial é, muitas vezes, inespecífica, apresentando-se como fadiga ou emagrecimento não intencional, o que gera, frequentemente, um grande atraso no diagnóstico. Os sintomas orgânicos específicos, por sua vez, permitem a identificação da doença, desde que exista uma alta suspeição clínica e investigação sistematizada.
|
Localização e frequência de acometimento |
Apresentação |
|
Rins (60 a 70%) |
Proteinúria nefrótica, hipoalbuminemia, dislipidemia, edema e derrames cavitários. |
|
Coração (70 a 80%) |
Principal causa de morte. Insuficiência cardíaca com fração de ejeção preservada (ICFEp). Baixa voltagem e bradiarritmias ao ECG. Espessamento ventricular concêntrico, disfunção diastólica e contração atrial deficitária ao ecocardiograma. Trombos atriais e complicações tromboembólicas. Elevação de troponina e BNP. |
|
Sistema nervoso |
Neuropatia de fibras finas com predomínio sensorial. Disfunção autonômica: distúrbios de motilidade gastrointestinal, síndrome sicca, hipotensão postural, disfunção erétil e bexiga neurogênica. Síndrome do túnel do carpo bilateral. |
|
Boca |
Macroglossia (10 a 20%) e aumento das glândulas salivares. |
|
Trato gastrointestinal |
Acalásia, síndrome de mau esvaziamento gástrico, pseudobstrução intestinal, supercrescimento bacteriano do intestino delgado e sangramento gastrointestinal. |
|
Fígado e baço |
Hepatomegalia, colestase e hiperbilirrubinemia. Hipoesplenismo. |
|
Sangue |
Propensão aos sangramentos e equimoses, como as típicas periorbitárias. Deficiência do fator X por mecanismos diversos. |
|
Pele e anexos |
Distrofia ungueal e alopecia. |
|
Sistema locomotor |
Artropatia amiloide, sinal da almofada dos ombros e ruptura espontânea do tendão do músculo bíceps braquial. |
Como pode ser feito o diagnóstico da amiloidose AL?
O diagnóstico da amiloidose AL deve ser sempre considerado nos seguintes cenários: proteinúria não explicada, cardiomiopatia restritiva, neuropatia periférica com disfunção autonômica, síndrome do túnel do carpo bilateral, hepatomegalia sem causa clara, gamopatia monoclonal ou mieloma múltiplo com manifestações atípicas, como a macroglossia e equimoses periorbitárias espontâneas.
A confirmação diagnóstica exige a combinação dos seguintes elementos: identificação tecidual de depósitos amiloides (por meio do famoso padrão de birrefringência esverdeada com a coloração pelo corante vermelho do Congo à luz polarizada), seja no órgão acometido ou nos substitutos mais facilmente acessíveis (gordura subcutânea — com sensibilidade de até 75%, glândulas salivares menores, gengiva, reto ou pele) e identificação de discrasia sanguínea plasmocitária.
A avaliação simultânea da gordura abdominal e medula óssea permite a identificação de depósitos amiloides em 85% dos casos de amiloidose AL.
A presença de proteína monoclonal pode ser evidenciada por meio das seguintes ferramentas: (1) imunofixação de proteínas séricas e urinárias, (2) pesquisa de cadeias leves séricas (free light) ou (3) identificação de plasmócitos com produção exclusiva de cadeias lambda ou kappa na medula óssea.
Uma vez definida a amiloidose AL, a realização do mielograma e biópsia da medula óssea são imprescindíveis para avaliar a carga de células plasmocitárias e excluir diagnósticos concomitantes, como o mieloma múltiplo e doenças linfoproliferativas envolvendo as células B, como a leucemia linfocítica crônica, linfoma indolente e macroglobulinemia de Waldenström.
Outro órgão-alvo a ser avaliado sistematicamente é o coração, por meio de ecocardiograma com doppler (com destaque para a modalidade strain), a ressonância magnética cardíaca, o PET-TC utilizando o radiotraçador 18F-florbetapir e a cintilografia óssea.
É possível definir qual o precursor proteico envolvido?
A definição do precursor proteico é essencial para nortear o tratamento. A identificação pode ser realizada em laboratórios experientes e com painel amplo de anticorpos através de estudos imunohistoquímicos, microscopia eletrônica ImmunoGold e a espectrometria de massa, essa última considerada a abordagem de maior acurácia, com sensibilidade de 88% e especificidade de 96%.
Como podemos estagiar a doença e avaliar o seu risco evolutivo?
A evolução da amiloidose AL dependente, fundamentalmente, da gravidade do acometimento cardíaco ao diagnóstico, que quando avançado associa-se a sobrevida média de 3 a 6 meses. Em contrapartida, na ausência de acometimento cardíaco, o paciente pode sobreviver muitos anos.
Elencamos, aqui, alguns importantes parâmetros de mau prognóstico, com os seus respectivos pontos de corte entre os parênteses:
- BNP > 81 a 700 pg/mL;
- NT-pró-BNP > 332 a 8.500 pg/mL;
- Troponina > 0,025 a 0,1 ng/mL;
- Taxa de filtração glomerular estimada < 50 mL/min/1,73 m²;
- Proteinúria em 24 horas (> 5 g);
- Diferença entre cadeias leves circulantes envolvidas e não envolvidas > 180 mg/dL.
Outros parâmetros prognósticos existentes, mas não incorporados aos sistemas de estadiamento tradicionais, são o fator de von Willebrand, d-dímero e fator de diferenciação de crescimento 15.
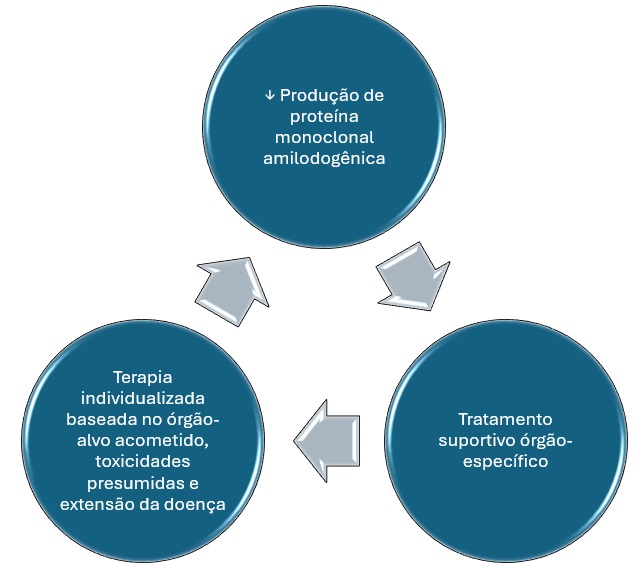
Quais são os pilares terapêuticos da amiloidose AL?
Os três principais pilares do tratamento da amiloidose AL encontram-se sintetizados na figura abaixo.
|
Disfunção orgânica |
Tratamentos suportivos disponíveis |
|
Miocardiopatia amiloide |
Restrição sódica; Diureticoterapia parcimoniosa; Vasodilatação (iECA); Antagonistas do receptor mineralocorticoide; Cuidados adicionais com anticoagulação (risco aumentado de sangramentos); Evitação de bloqueadores de canais de cálcio; Marcapasso (síncope relacionada a bradiarritmias); Cardiodesfibrilador implantável (TV/FT abortadas); Dispositivos de assistência ventricular; Transplante cardíaco. |
|
Hipotensão postural |
Fludrocortisona, em geral, não é uma boa opção, pela retenção hidrossalina. Meias elásticas compressivas; Midodrina, piridostigmina ou droxidopa. |
|
Nefropatia amiloide |
Restrição sódica; Diureticoterapia; Manejo da dislipidemia; iECA/BRA (proteinúria); iSGLT2 (estudos em andamento nesse cenário); Hemodiálise; Diálise peritoneal. |
|
Diarreia |
Loperamida ou difenoxilato-atropina; Octreotide; Tratamento de supercrescimento bacteriano do intestino delgado. |
|
Dor neuropática |
Gabapentina, pregabalina e duloxetina. |
|
Deficiência adquirida do fator X |
Esplenectomia; Reposição de fatores de coagulação. |
Qual é o tratamento específico de primeira linha na amiloidose AL?
O tratamento específico da amiloidose é direcionado à supressão da produção da proteína monoclonal.
A semelhança dos tratamentos instituídos para o mieloma múltiplo, encontram-se disponíveis:
- Melfalan + transplante autólogo de células tronco hematopoiéticas (TMO), tratamento reservado aos pacientes com bom status performance e sem doença multiorgânica avançada (menos de 20% do total), combinação capaz de induzir resposta hematológica completa em 40% dos indivíduos.
- CyBorD (Ciclofosfamida, Bortezomibe, Dexametasona e Daratumumabe): tratamento ofertado para os pacientes inelegíveis ao TMO, com resposta parcial ou melhor em quase 80% dos casos.
Aos pacientes com recidiva ou progressão da doença a despeito da terapia inicial, as principais terapias de segunda linha incluem a repetição do tratamento de primeira linha, caso o mesmo tenha desencadeado resposta com duração mínima de 1 ano; inibidores do proteassoma, anticorpos monoclonais anti-CD8, imunoterapia, venetoclax nos casos de t(11;14), bendamustina, TMO + melfalan e terapia CAR-T cell.
Existem tratamentos capazes de reabsorver e degradar os depósitos amiloides?
Dois anticorpos antifibrilares, birtamimabe e anselamimabe, estão sendo investigados para a remoção de fibrilas amiloides de órgãos por meio da ativação do sistema imune para a degradação química e enzimática, seguida por fagocitose mediada por anticorpos.
No estudo fase 3 VITAL, o birtamimabe demonstrou benefício de sobrevida entre pacientes com amiloidose cardíaca AL avançada em estágio IV.
Avaliação da resposta terapêutica
Os parâmetros de resposta hematológica e orgânica completa incluem:
- Ausência de proteína monoclonal sérica e urinária;
- Relação preservada de cadeias leves séricas (ou preponderância das cadeias leves não acometidas);
- NT-pró-BNP ≤ 250 pg/mL ou BNP ≤ 80 pg/mL;
- Proteinúria de 24 horas ≤ 200 mg;
- Redução da fosfatase alcalina ≤ 50%;
- Redução do tamanho hepático maior ou igual a 2 cm.
A resposta hematológica, quando precoce e profunda, se associa à sobrevida prolongada, sendo acompanhada por melhora orgânica, sendo que a última pode demorar de 6 a 24 meses para se tornar perceptível, e com alcance da melhor resposta somente em 36 meses em alguns casos, a exemplo do envolvimento hepático.
Conclusão e mensagens práticas
- A amiloidose AL é uma doença grave e rapidamente progressiva, com prevalência crescente nas últimas décadas.
- O arsenal terapêutico, realocado a partir de terapias destinadas ao mieloma múltiplo, tem revolucionado a sua abordagem, com incremento da sobrevida em 5 anos de 15 para 50%.
- A condição deve ser suspeita de proteinúria não explicada, cardiomiopatia restritiva, neuropatia periférica com disfunção autonômica, síndrome do túnel do carpo bilateral, hepatomegalia sem causa clara, gamopatia monoclonal ou mieloma múltiplo com manifestações atípicas.
- O diagnóstico é confirmado pela identificação tecidual de depósitos amiloides e discrasia sanguínea plasmocitária.
- O transplante autólogo de medula óssea, associado ao Melfalan; e o esquema CyBorD são os principais tratamentos destinados a esse grupo de pacientes.
Autoria

Leandro Lima
Editor de Clínica Médica da Afya ⦁ Residência em Clínica Médica (2016) e Gastroenterologia (2018) pelo Hospital das Clínicas da Universidade Federal de Minas Gerais (HC-UFMG) ⦁ Residência em Endoscopia digestiva pelo HU-UFJF (2019) ⦁ Preceptor do Serviço de Medicina Interna do HU-UFJF (2019) ⦁ Graduação em Medicina pela Universidade Federal de Juiz de Fora (UFJF)
Prescreva medicação aos seus pacientes de forma gratuita e ilimitada
Como você avalia este conteúdo?
Sua opinião ajudará outros médicos a encontrar conteúdos mais relevantes.